O-Mannosylation
O-mannosylated glycans have first been described on glycoproteins of the yeast cell wall. This type of O-glycosylation is twice as frequent as N-glycosylation on cell wall glycoproteins and is essential for the viability of the yeast cell. Surprisingly, the first report of O-mannosylated glycans in mammals only dates back to 1997 when the group of Tamao Endo described such glycans on the adhesion protein α-dystroglycan. Soon after this pioneer work, O-mannosylation was recognized in all animals, and demonstrated to be essential for muscular adhesion and neuronal migration processes.
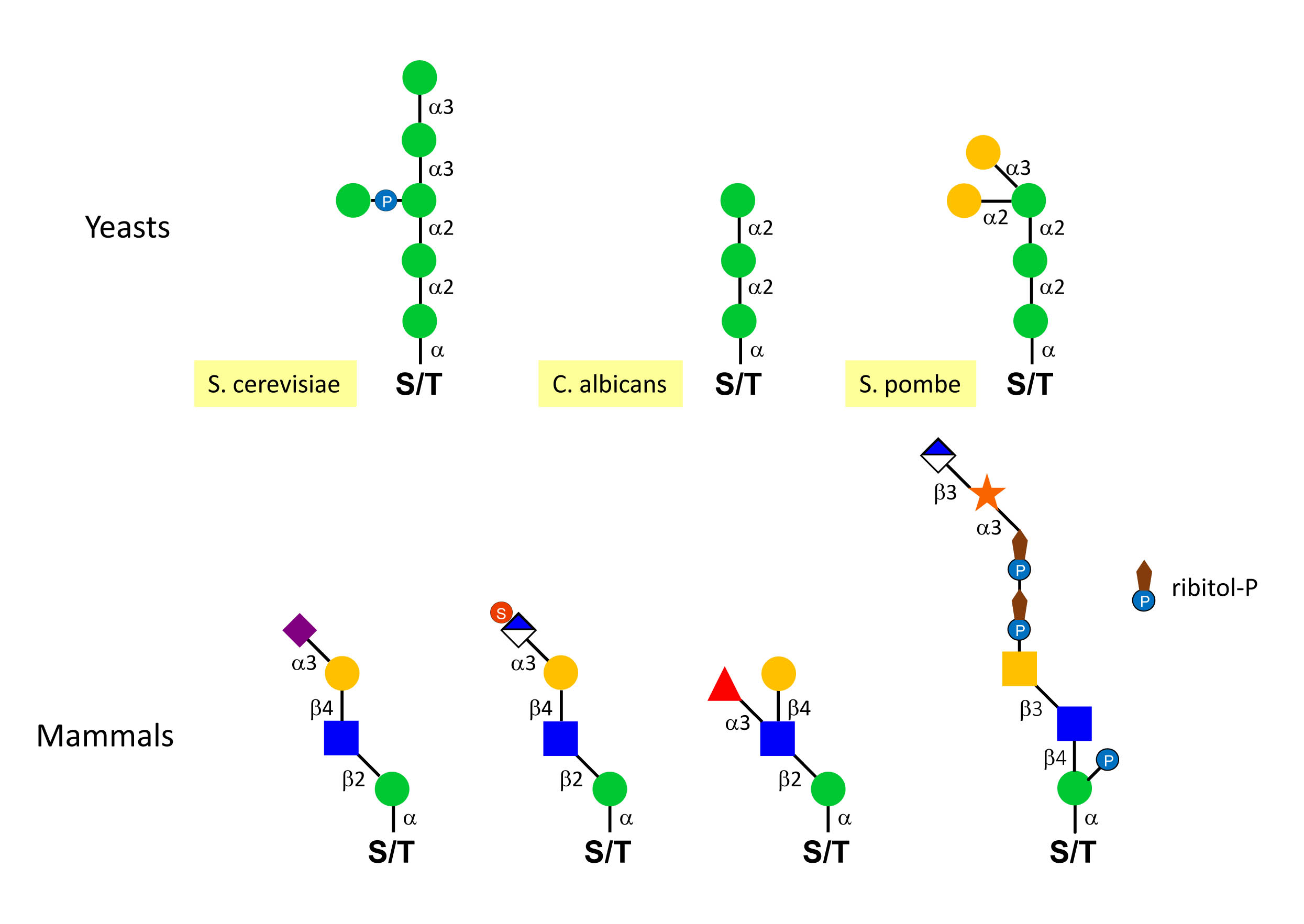
The structures of O-mannosylated glycans are radically different between yeast and higher eukaryotes. Yeast O-mannosylated glycans are mainly linear chains of Man units with some species-specific variations, such as the addition of Man-P as a side chain in Saccharomyces cerevisiae or terminal α-linked Gal in Schizosaccharomyces pombe. Higher eukaryotes like mammals display more structural variability, including different core structures terminated by various glycans such as the HNK-1 epitope (SO4-GlcA[β1-3]Gal[β1-4]GlcNAc) and long repeats of the disaccharide GlcA[β1-3]Xyl[α1-3 ] that are catalysed by the LARGE glycosyltransferase. The LARGE protein is in fact a double glycosyltransferase including an α1-3 Xyl-transferase domain and a β1-3 GlcA-transferase, which assemble a polymeric chain of GlcA[β1-3]Xyl[α1-3 ]. LARGE can also elongate mucin-type O-GalNAc glycans, which are also found on α-dystroglycan.
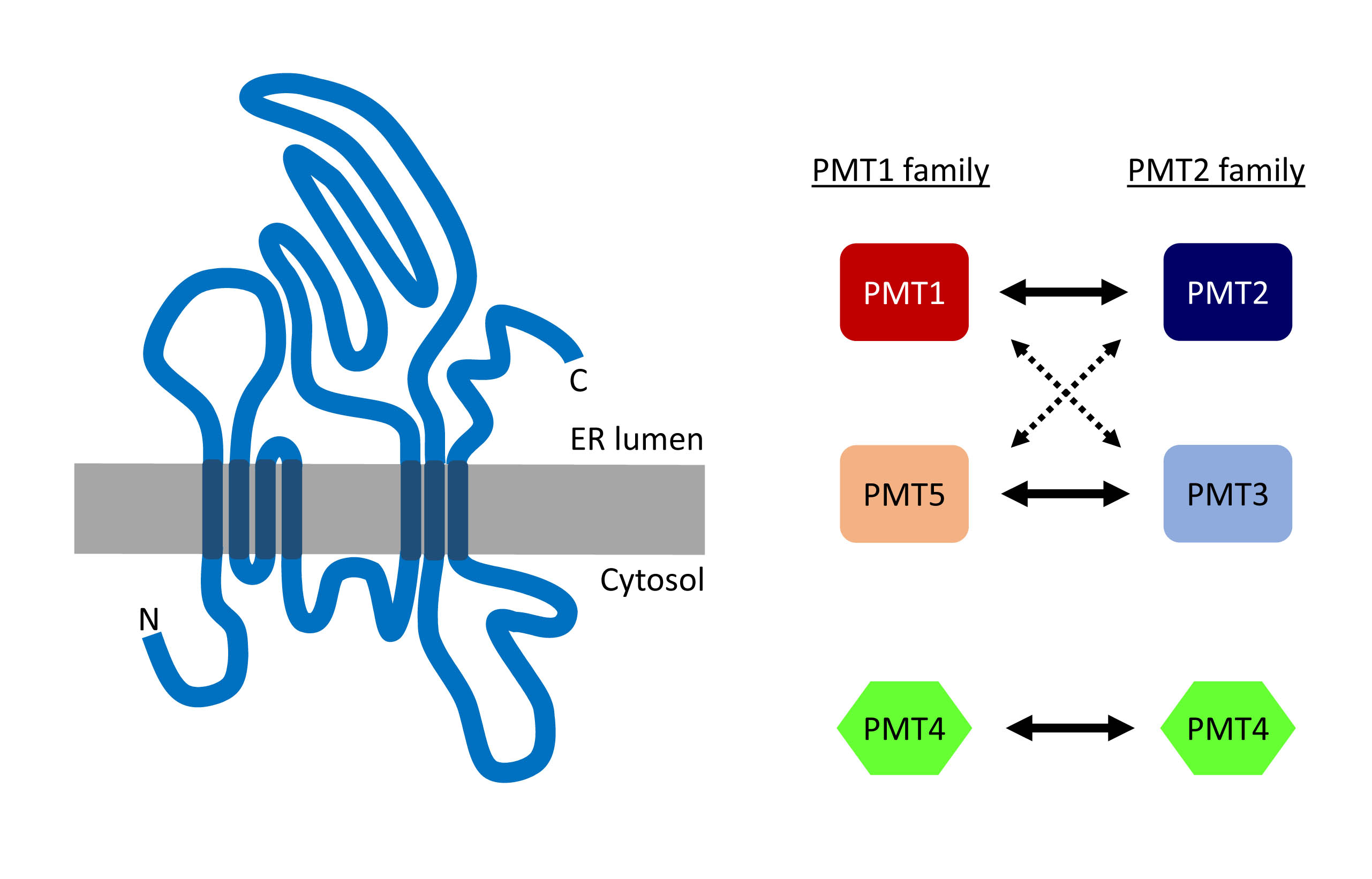
The transfer of Man to serine and threonine takes place in the ER and is catalyzed by the family of protein o-mannosyltransferases, abbreviated PMT in fungi and POMT in higher eukaryotes. The yeast Saccharomyces cerevisiae expresses six PMT proteins, which can be grouped in three structurally subfamilies. By contrast, only two POMT proteins are found in higher eukaryotes. PMT/POMT enzymes have multiple trans-membrane domains spanning the ER membrane. Several loops extending in the ER lumen are likely to be important for the binding to the acceptor glycoproteins but the exact topology and reaction mechanisms are still unknown. Because PMT/POMTs use Dol-P-Man as donor substrate, their hydrophobic regions are also involved in the catalysis of the Man transfer. PMT/POMT enzymes are only active as dimers, mainly heterodimers but also homodimers in the case of Saccharomyces cerevisiae PMT4. In higher eukaryotes, POMT1 and POMT2 also need to build heterodimers to be active.
O-mannosylation is essential for yeast viability, although the exact roles of O-Man glycans are still unclear. O-mannosylation increases the solubility of secreted proteins, which can contribute to the folding and secretion of essential cell wall proteins. In animals, the first functional assignment for O-mannosylation occurred in Drosophila with the description of the two mutations rotated abdomen (ra) and twisted (tw). These mutations cause a defect of muscular development, which is characterized by a staggering of abdominal segments. The link between these mutations and O-mannosylation was made by noticing the sequence similarity of the ra and tw proteins with the yeast PMT enzymes. As confirmed later, the ra and tw proteins are in fact the Drosophila orthologs of POMT1 and POMT2.
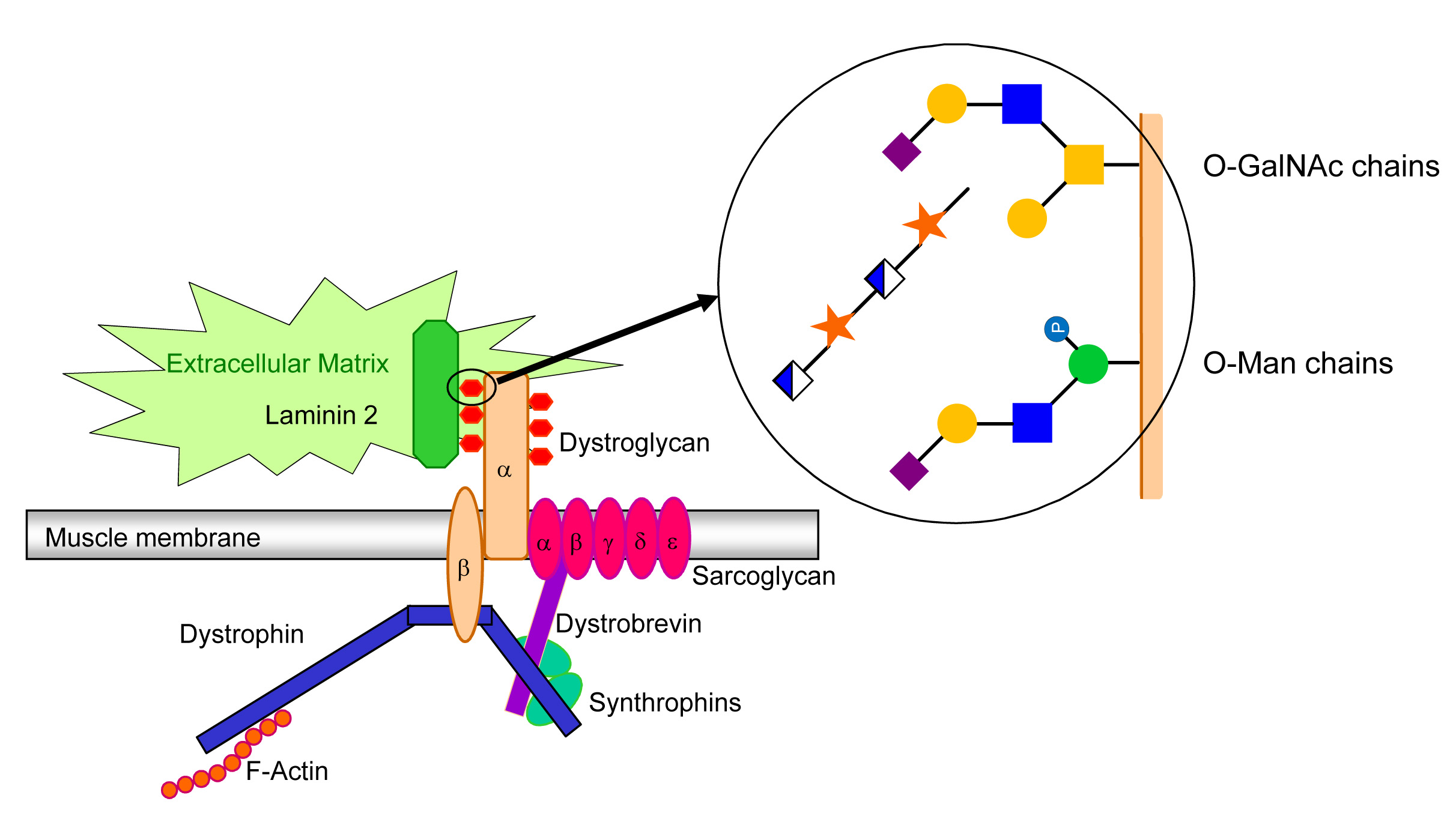
Despite their ubiquity in eukaryotes, O-Man glycans have only been found on few glycoproteins in higher eukaryotes. The best studied of these glycoproteins is α-dystroglycan, a large glycoprotein of 150 kDa, of which glycan chains account for nearly 100 kDa. α-Dystroglycan carries multiple O-Man and O-GalNAc glycans, which are essential for the binding of α-dystroglycan to its ligands in the extracellular matrix. For example, α-dystroglycan binds to laminin-2, which is important for the adhesion of muscle cells and hence for muscular stability. Accordingly, defective dystroglycan-laminin interactions lead to muscular degeneration, which is the hallmark of congenital muscular dystrophies. The dystroglycan-laminin complex is also relevant in the context of infectious diseases. In fact, Mycobacterium leprae infects Schwann cells after docking on dystroglycan-laminin. The O-Man glycans on α-dystroglycan also function as receptor for the lymphocytic choriomeningitis virus and Lassa fever virus.
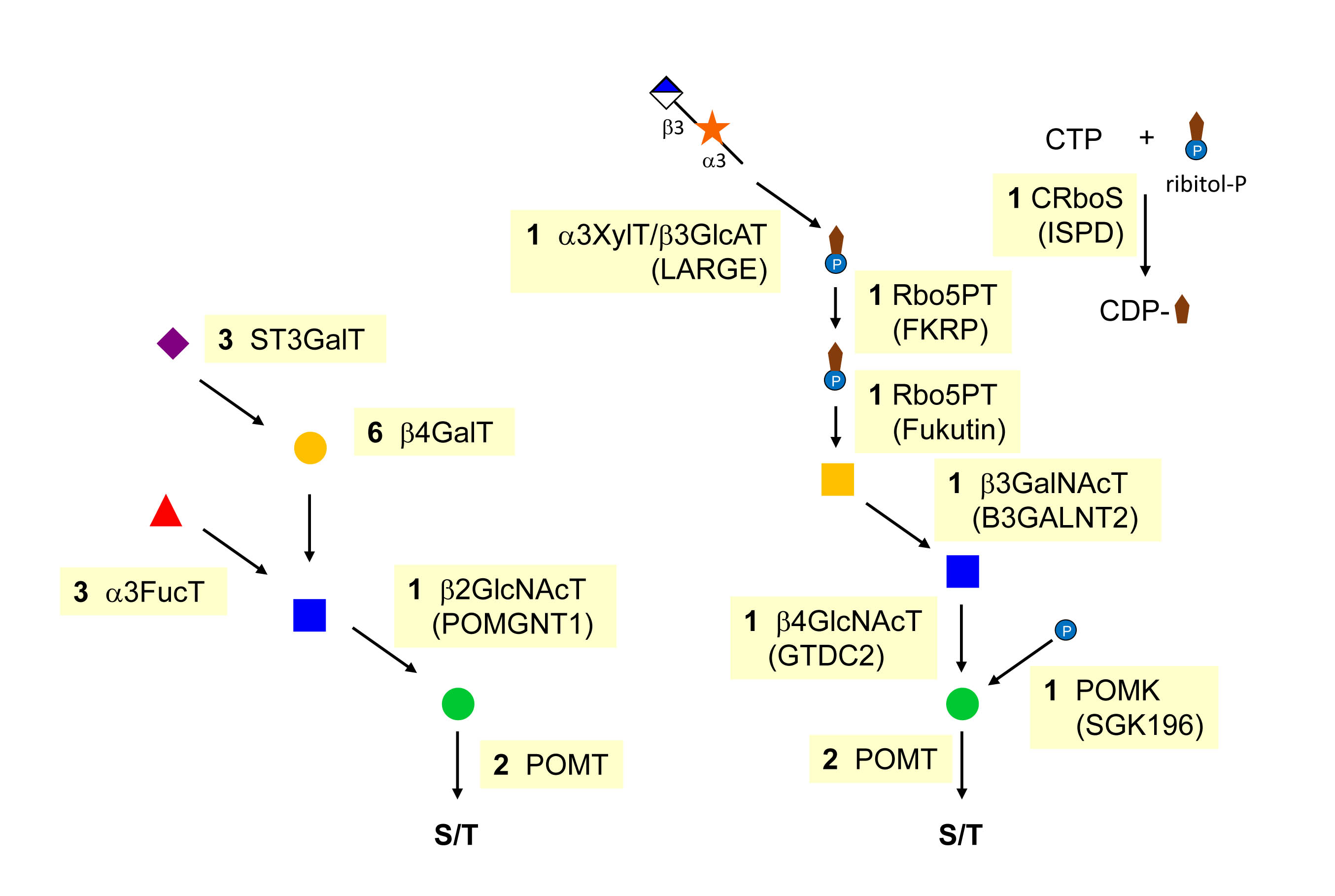
O-Mannosylation was long considered to be a fungi-specific form of O-glycosylation and very few would have predicted that a large family of diseases could be related to this type of glycosylation. The first of these diseases was reported 1993, when several groups identified mutations in Fukutin gene as a cause of Fukuyama-type congenital muscular dystrophy. Other types of congenital muscular dystrophies were later associated with additional putative glycosyltransferases involved in O-mannosylation. Deficiency in POMT1, POMT2, POMGNT1, POMGNT2 (GTDC2), B3GALNT2, SGK196, LARGE, Fukutin and Fukutin-related protein (FKRT), TMEM5, and isoprenoid synthase domain-containing protein (ISPD) have been associated with the Walker-Warburg syndrome, Muscle-Eye-Brain disease, Fukuyama-type congenital muscular dystrophy and Limb Girdle muscular dystrophy. Interestingly, the enzymes ISPD, Fukutin and FKRT are involved in the biosynthesis and transfer of ribitol-5-P, a pentose alcohol that is a constituent of teichoic acid found in the cell wall of Gram-positive bacteria. Sugar alcohols are otherwise unknown in animal glycans. Ribitol-5-P is generated by reduction of ribose and activated to CDP-ribitol through the action of ISPD. Fukutin and FKRP are two ribitol-5-P transferases that use CDP-ribitol as donor substrate.
Besides its contribution in maintaining muscular integrity, α-dystroglycan is also involved in the migration of cortical neurons and development of retinal tissue. The dystroglycan-laminin complex enables the formation of a barrier called glia limitans, which prevents neurons from migrating into the subarachnoid space. Without glia limitans, neurons overshoot across the marginal zone, which leads to structural abnormalities hinted to in the name of the corresponding disease: “cobblestone” lissencephaly. Patients featuring defects of O-mannosylation are severely mentally retarded and often blind.
Although mutations in the core glycosyltransferase genes POMT1 and POMT2 are often associated with the most severe forms of congenital muscular dystrophies like the Walker-Warburg syndrome, the severity of the disease is more related to the level of residual enzymatic activity rather than to the position of the mutated gene along the glycosylation pathway (i.e. core vs. terminal glycosyltransferase). Accordingly, mutations in all glycosylation genes involved in O-Man glycan biosynthesis have been associated with severe forms of congenital muscular dystrophy.