Diseases of ER N-glycosylation
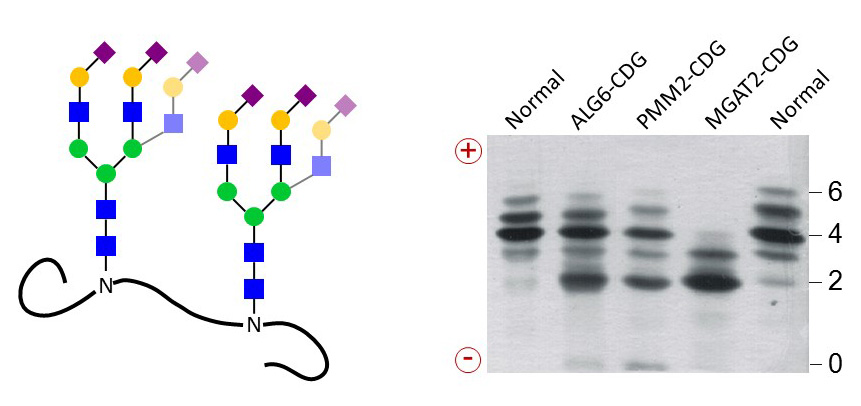
Defects in the assembly of Dol-linked oligosaccharides decrease the pool of substrates for the N-linked glycosylation of nascent proteins. Such glycosylation defects can be diagnosed by isoelectric focusing (IEF) of blood serum transferrin, which carries two N-glycans. IEF separates the glycoforms of transferrin by negative charges, which are carried by terminal Sia on N-glycans. The simplicity of this test was instrumental in identifying more than 50 N-linked glycosylation defects in the last twenty years.
Figure 35. Serum transferrin carries two N-glycans normally terminated with four to six negatively charged Sia residues. Separation of transferrin glycoforms by IEF yields bands for each number of Sia. Transferrin from CDG patients lack complete N-glycans or only terminal carbohydrates, thereby decreasing the number of negative charges.
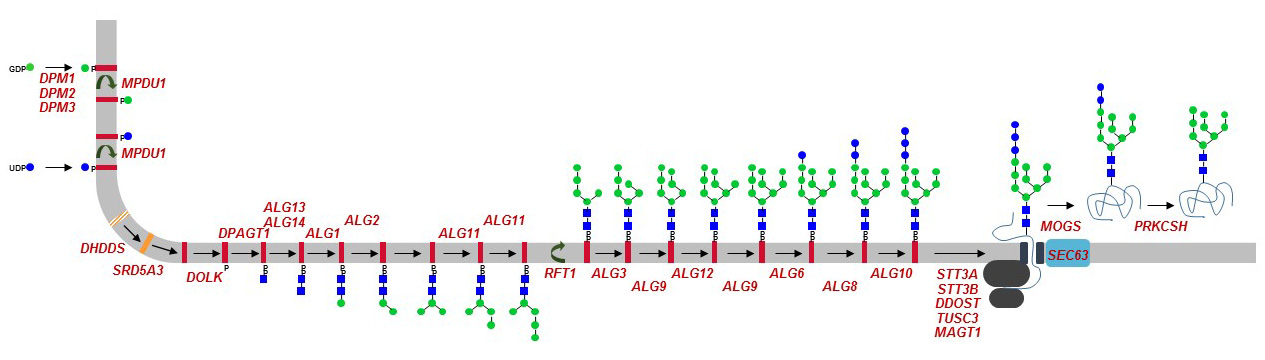
Nearly all genes involved in the biosynthesis of Dol, GDP-Man, Dol-P-Man and of Dol-linked oligosaccharides have been identified as cause of a glycosylation defect in humans. Mutations in some OST subunit genes and in the glucosidase-I and –II genes have also been described. Because of their similar yet broad clinical manifestations, defects of N-glycosylation have been grouped under the general term of congenital disorders of glycosylation (CDG). Soon after the description of the first cases, CDG have been divided into two groups, where CDG type-I, or CDG-I in short, encompassed glycosylation defects leading to the non-occupancy of N-glycosylation sites on glycoproteins, and CDG-II encompassing all other glycosylation defects. Accordingly, most ER N-glycosylation defects belonged to CDG-I. Individual gene defects were further specified by adding a distinct letter, thereby yielding CDG-Ia, CDG-Ib, CDG-Ic, etc. The identification of mutations affecting multiple glycosylation pathways prompted a revision of the nomenclature to a more open system. Therefore, CDG are currently designated by simply linking the HGNC approved symbol of the mutated gene to the suffix “CDG”, thus defining PMM2-CDG, ALG6-CDG, and DPM1-CDG.
{kind=link}
Figure 36. ER N-glycosylation defects identified in CDG patients. Defective genes are marked in red. The overview of all glycosylation defects known to date can be downloaded here:
Neurological alterations are prominent in defects in ER N-linked glycosylation. Typical symptoms include psychomotor retardation, ataxia, hypotonia, epilepsy, and visual impairments. Endocrine disorders, coagulopathy, gastrointestinal and hepatic impairments, cardiomyopathy, are though also frequently observed. The severity of the diseases is variable and depends largely on the level of residual glycosylation enabled by individual gene defects. Most severe impairments lead to infant lethality and weak mutations lead to moderate intellectual delays.
Defective phosphomannomutase activity is the most frequent form of CDG in respect to the number of mutations in the PMM2 gene and number of patients identified to date. The PMM2 enzyme catalyzes the conversion of Man-1-P from Man-6-P, which is an essential reaction in the biosynthesis of GDP-Man. Unfortunately, most CDGs remain without therapy, with the exception of mannose phosphate isomerase (MPI) defect, which can be circumvented by oral Man supplementation. MPI mediates the interconversion of fructose-6-P and Man-6-P, an important step in the maintenance of the cellular Man pool. MPI-CDG lacks the typical neurological symptoms of CDG and rather features gastrointestinal defects including diarrhea, protein-losing enteropathy, and intestinal bleeding.