Diseases of Golgi N-glycosylation
Only few defects of Golgi N-glycosylation are known in humans to date. Some are asymptomatic like the deficiencies of the terminal fucosyltransferases FUT3 and FUT6, which are found in about 9% of individuals on the island of Java in Indonesia. This lack of apparent phenotype is surprising considering that FUT6 is a major fucosyltransferase involved in the fucosylation of liver proteins.
A single defect of N-glycan branching has been identified to date. Mutations in the MGAT2 gene lead to the loss of complex N-glycans. This defect causes a severe disease, characterized by multiple organ dysfunctions and elevated childhood lethality. A rare form of galactosylation deficiency is caused by mutations in the β1-4 Gal-transferase B4GALT1 gene. So far only two patients have been identified with B4GALT1-CDG. In the first case, B4GALT1 deficiency was associated to severe clinical features such as psychomotor retardation, hypotonia, myotonia and macrocephaly. Paradoxically, the second case presented with mild symptoms of mental retardation and hepatic dysfunction. It is currently unclear whether the second case represented only a partial galactosylation defect.
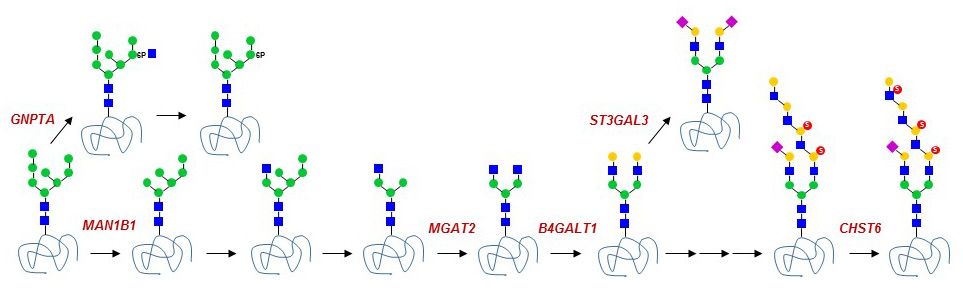
Figure 41. Golgi glycosidase and glycosyltransferase defects known to date.
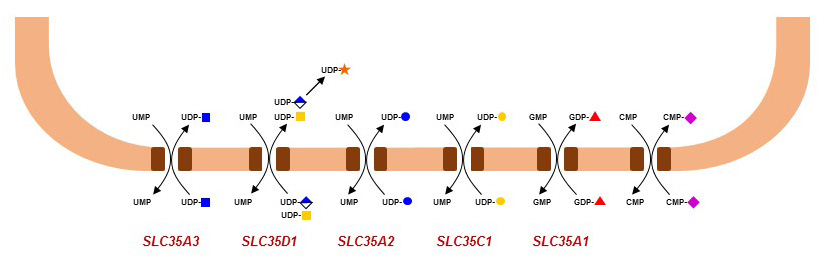
Glycosylation defects in the Golgi are usually not specific to N-glycans, because enzymes such as B4GLAT1 also act on several types of O-glycans and glycosphingolipids. The same remark applies to defects of nucleotide-activated sugar transport into the Golgi, since these donor substrates are used to elongate multiple types of glycans. For example, mutations in the SLC35C1 gene encoding the Golgi GDP-Fuc transporter leads to a generalized defect of fucosylation in multiple classes of glycans. By contrast, ER O-fucosylation is not affected by the SLC35C1 defect, indicating that another GDP-Fuc transporter is active in the ER. The loss of Golgi fucosylation leads to a form of immunodeficiency called Leukocyte Adhesion Deficiency type-II (LAD2) or SLC35C1-CDG. Fucosylated glycans, such as sialyl Lewis-X, are ligands of selectin proteins, which are important for the rolling of leukocytes on blood vessel walls and thereby for the extravasation of leukocytes into tissues. The treatment of LAD2 patients with dietary Fuc has been shown to partially restore Golgi fucosylation and leukocyte rolling. Mutations in the CMP-Sia acid transporter gene SLC35A1 have also been described in two infants presenting distinct pathologies. The first child was affected by with recurrent bleeding episodes, thrombocytopenia, neutropenia and multiple bacterial infections, whereas the second child presented with intellectual impairment, seizures, ataxia, renal and cardiac disorders. Additional cases are required to delineate the importance of sialylation in human development and physiology.
{kind=link}
Figure 42. Golgi-localized nucleotide-activated sugar transporter defects known to date.